A new site that serves as the first knowledge base of its kind in Hebrew in the field of rare genetic diseases in Israel - was launched on the occasion of the International Day for Rare Diseases, which is noted on a date that only returns once every four years - February 29. The Internet project is a joint initiative of Bar-Ilan University and the Weizmann Institute of Science
According to an estimate, in Israel there are about half a million children diagnosed with rare genetic diseases. According to Dr. Einat Salzkaber, a researcher at the Faculty of Life Sciences at Bar-Ilan University, despite the large number, parents of children diagnosed with a rare disease feel terribly alone. "The reason for this is that the rare diseases are so many and different, and with the diagnosis parents find themselves struggling for every piece of relevant information that is usually not accessible to all researchers and doctors, let alone to the general public. The parents do not know where to turn, which doctor, which association. Even those involved in rare disease research do not always know each other, or are aware of research and developments in the field. Considering the estimate that there are about 7,000 different rare diseases, it is clear that no one can specialize in all of them."
For this reason, Dr. Salzkaber, and Prof. Ayelet Erez and Prof. Maya Shuldiner, from the Weizmann Institute of Science, founded the website "Rare genetic diseases in Israel", Used as a first-of-its-kind online database in Hebrew. The database allows researchers, doctors, biotech companies and patients' families to update and be updated. Carmel Liebman, translates the disease values and makes the information accessible to the various agencies.
The connections created by the site between the various parties are of the utmost importance as they not only make information accessible to patients and their families, but create collaborations between experts, associations and rare families that lead to research breakthroughs and the development of life-saving treatments. The site, which was established at the Weizmann Institute of Science with funding from the Center for the Research of Hereditary Diseases for the David and Pela Chappel Family, already includes thirty disease entries and a large number of entries from researchers, doctors and patient associations. The ambition is that in the future it will include at least one hundred common rare genetic diseases in Israel, which will cover over 90% of the rare population.
On the occasion of this year's Rare Disease Day, this year the Weizman Institute and Bar-Ilan chose to publicize the establishment of the site and promote awareness of rare diseases in a unique art project: called "Rare". As part of the project, 11 artists each created an original work inspired by a rare genetic disease, each from their unique point of view and with their characteristic technique.
"It is impossible to imagine what we will be able to give these patients in five or ten years, in terms of diagnosis and treatment," says Dr. Salzkaber. "Science is advancing in giant strides, so it is critical that the Israeli academy join forces for the sake of the patients and their families, and give them real hope."
the works

Rett Syndrome - Hadar Zisman
Maximum incidence in the population: 1:10,000 births of girls
A syndrome characterized by progressive muscular atrophy and is mainly common among girls. The patients suffer from a variety of disorders such as the loss of the ability to speak and use the hands, walking disorders, irregular breathing and digestive difficulties.
Hadar Zisman was diagnosed with Rett syndrome at the age of two and a half. Today, at the age of twenty, she is a student in a special undergraduate program at Bar-Ilan University. Her father, Shmulik, is the founder of the "Angels of Silence" association, and thanks to the High Court of Justice filed on her behalf, a gaze targeting system was introduced to the healthcare basket in Israel, which allows girls with the syndrome to communicate with the world. Her piece in the project - entitled "How is it not to speak?" - Hadar drew with her eyes.

STXBP1 syndrome
Tali Hivrich
Maximum prevalence in the population: 1:30,000
A syndrome resulting from a deficiency in the normal STXBP1 protein in the cells, which is accompanied, in most cases, by various types of convulsions that may appear already in the first days of the children's lives. Despite the brain and nerve damage, which causes difficulties in motor function and the loss of the ability to speak, the sick children smile and laugh.
Maple Syrup Syndrome (MSUD)
Galia Haley Pasternak
Maximum prevalence in the population: 1:185,000
A severe recessive hereditary disease. It is so named because the first detectable clinical sign within 12 hours of birth is urine/earwax that smells like maple syrup. Diagnosing the disease at birth may save the baby's life, which is why it is tested in the newborn screening that most babies in Israel undergo. The disease causes neurological deterioration and metabolic acidosis, and in cases where it is not diagnosed in time, may also lead to death.
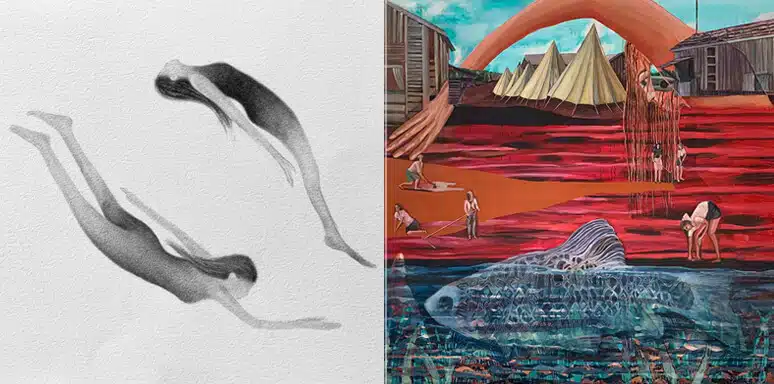
Cystic fibrosis
These are Amitai Sadovsky
Maximum prevalence in the population: more than 100,000 patients worldwide
One of the most common hereditary diseases in the West, and it is especially common among Ashkenazi Jews due to a specific ancestral inherited mutation. It affects the lungs, pancreas, digestive system and more. Lung disease is the main cause of morbidity and mortality in CF patients. In addition, the patients with the disease (and especially patients) tend to suffer from aquagenic wrinkles of the palms, a rare dermatological phenomenon that occurs in contact with water.
Marfan syndrome
Reut Dafna
Maximum prevalence in the population: 1:5,000-10,000
Marfan syndrome manifests itself mainly in the cardiovascular, skeletal and ocular systems. Most patients have a dilatation (aneurysm) of the aorta, and a typical unique appearance characterized by long limbs and prominent sternum.

phenylketonuria (PKU)
Efrat Yemin
Maximum prevalence in the population: 1:5,000-10,000
A genetic metabolic disease that, if not diagnosed and treated in time, causes developmental intellectual disability, epilepsy and autism. Patients with phenylketonuria are often characterized by light skin, eyes and hair. They must maintain a strict diet - they must not consume drinks containing the artificial sweetener aspartame, as well as foods high in protein, such as meat, dairy products and legumes, and they are also limited in the consumption of foods high in starch.
spinal muscular atrophy (SMA)
Amir Tumashov
Maximum prevalence in the population: 1-2:100,000
A serious disease, which causes progressive muscle weakness as a result of neurodegeneration. Following the struggle of parents of children with the disease, a drug was found that is considered the most expensive in the world and costs about two million dollars for one treatment. Recently, two drugs to treat the disease entered the medicine basket in Israel.

X-ALD
Photo: Neta Mozes. Glass sculpture: Michal Herda
Maximum prevalence in the population: 1:14,000-17,000
Hereditary disease of the nervous system, transmitted from a white mother and common among boys. It is characterized by attention and hearing disorders, up to the loss of hearing, vision and the ability to speak, which are caused by worsening neurological degeneration. The patients' chances of survival are low, but the deterioration of the disease can be slowed down by increased consumption of fats
not saturated The story behind the attempt to find a treatment that would slow down the progression of the disease, by the parents of a sick child, was the inspiration for the movie "Lorenzo's Oil".
Prader Willi (Prader Willi)
Dana Barlev
Maximum prevalence in the population: 1:10,000-30,000 is also called "fatal hunger syndrome". In early childhood, patients are characterized by excessive eating and the gradual development of morbid obesity, unless food intake is strictly controlled. Obesity results from hyperphagia, which is an extreme urge to consume food that is insatiable. Parents of sick children are forced to lock all the kitchen cabinets and the refrigerator.

Curse of Ondine (CCHS)
Tamar Lev-on
Maximum prevalence in the population: 1:148,000 births
Sleep apnea syndrome. During waking hours, the children with the disease are normal children in everything, but during sleep they stop breathing independently. The syndrome is named after the mythological nymph Undine, who according to legend cursed her unfaithful partner with a curse that would make him stop breathing every time he fell asleep.
Neurofibromatosis (NF1)
Philscher Foundation
Maximum prevalence in the population: 1:2,052
Patients with the disease may suffer from damage to the skeleton, learning difficulties and damage to the nervous system characterized by cognitive decline, epilepsy and more. The disease is characterized by a large number of skin spots called "coffee-in-milk" spots and appear in almost all patients in infancy and childhood. In the first years of life, the number of spots increases and so does their size, but they blur in older patients.
More of the topic in Hayadan:
- An autoimmune disease has been discovered that affects the development of tooth enamel and is common among children with celiac disease
- How a single gene that causes a rare disease helps in understanding complex diseases like cancer
- The genetic defect that causes a rare disease called "SOFT syndrome" has been revealed
